This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
By offering comprehensive and audit-proof documentation, the platform aims to simplify regulatory approvals and reduce time-to-market for pharmaceutical companies. The product database was developed to provide direct online access to all registration-relevant information.
Introduction Once, I received a query from a ward nurse: If a product label mentioned that "Do not store over 25°C", is it fine for us to keep the medication in fridge? What does a cool place on the product label mean? Read the label to know the recommended storage temperature range.
“It is not justifiable to compare pharma products, where a lot of work has already been done and well-documented with international harmonisation (e.g., “Ethical marketing focuses on education rather than exaggeration, empowering consumers with accurate information about product benefits.
” Main Purpose Regulatory application/ submission management A public database listing all FDA-approved small molecule drugs (not biologics) and providing information about equivalence, patents. Indian innovators would benefit from clear public information on patent life and exclusivity, incentivising high-value R&D investments.
This also allows patients to understand what medications they are taking and provide that information to other medical professionals if necessary. This is great as it allows prescribers to provide extra information about treatments and facilitates communications even when someone needs information out of hours.
6 ,7 One of the most critical ethical considerations is informed decision-making. 9 Labeling materials should describe how the technology works, its intended use, known limitations, and the circumstances under which human oversight is recommended. Also important is the need for clearly defined and delineated accountability.
Also, consistency across data sources should be taken into consideration; one should not fear having heterogeneous sources, yet it is crucial to afford the model a way to understand information in a cohesive and consistent way. This relates also to data discontinuity. There are pockets of very rich data and pockets of very sparse data.
Greater visibility into these rebate trends could enable oversight of affordability trends, foster more informed public discourse, and allow patients and advocates to better anticipate the evolving cost burden on the Medicare program. This will allow other stakeholders (e.g.,
Best practices include: Clearly stating the drug name, indications, and fair balance information. Avoiding misleading claims or unapproved off-label promotions. Using call-to-action (CTA) phrases such as Learn More, Request Information, or Get HCP Insights.
Scientific communication : Drafting slide decks, scientific responses, medical information letters, and training content using generative models, ensuring scientific accuracy and compliance in messaging and content. Clinical trials One reason clinical trials fail is choosing the wrong patients.
Image Credit: catalin | stock.adobe.com STARGLO Trial Overview Study Design and Population STARGLO was a global, randomized, open-label, phase 3 trial enrolling 274 patients across 62 centers in 13 countries. 7015 FDA Advisory Committee Briefing Document: Columvi (glofitamab-gxbm) plus GemOx. month follow-up was 13.5 (7.9-18.5)
This change mirrored a January 2025 FDA safety alert and label update that urged providers to consider genetic testing, inform patients of the risks, and discuss available options. Real-World Evidence, Emerging Therapies, and Demonstrating Value Technological advances are accelerating this progress.
The manufacturer reported that, as part of the NDA review, they have not highlighted any technical concerns related to the submitted documentation or testing of OLC. 2,4 The open-label, single-arm, multicenter, multidose study enrolled 86 patients with CKD and hyperphosphatemia receiving maintenance hemodialysis. 2024;35(10S).
Existing policies often feature explicit exclusions for non-FDA approved products or harm from off-label use. Maintain unwavering transparency with patients, unequivocally communicating the non-FDA approved status of compounded medications and all associated risks, securing explicit informed consent.
Stage 1: design the compounding blueprint Establish a complete plan including ingredients, packaging, calculations, labeling, and patient-specific factors. Finalize documentation with full traceability and ensure preparations are quarantined until verified.
Most medical documentation is polluted with it, for want of using the persons actual name. Subscribe to our email list to keep informed on all of the Resuscitation and Critical Care goodness. Facebook RSS Twitter YouTube CME Information See our CME Information Other Stuff Have a great idea for the next podcast?
The net payment made by Sino to acquire LaNova will be approximately $500.9m, a figure that excludes the estimated cash and bank deposits, according to a company document outlining the terms of the transaction. At the time, Sino spent 142 million yuan ($19.80m) to initiate its ownership involvement with the biotech.
Informed decision-making requires evidence grounded in scientifically validated methodologies, whether for clinical development, regulatory approval, or patient care. Without rigorous methods, transparency, and reproducibility, it’s information – but not evidence. This distinction matters.
One less utilized method (and for good reason) to gain more certitude is the 513(g) Request for Information. Before FDA can begin its review of a 513(g) Request for Information submission, the applicant must pay the user fee. For fiscal year 2025, which began on October 1 and runs through September 30, 2025, the standard fee is $7,301.
The guidance encourages sponsors to engage with FDA using the Q-Submission Program prior to submitting a PCCP in order to obtain FDA feedback on if the proposed modification is suitable for inclusion in a PCCP and what information the PCCP will need to include. FDA may request additional information during the review of the PCCP.
Intubated patients in the ED should have soft, wrist restraints places without the need for arduous, high-risk documentation or sitters Intubation Checklist EMCrit 176 – Updated EMCrit Rapid Sequence Intubation Checklist Awareness during Paralysis EMCrit 331 – Awareness after Resus RSI and ICU Paralysis – It is Unacceptable!!!
Both guidance documents recommend data management practices for collecting data for use in developing, tuning, and testing an artificial intelligence model and making changes to said model. If data are excluded because of data quality issues, the rationale and criteria for the exclusions should be documented in the DCP.
There are also drugs the Assessment identifies as being used off-label without high-quality evidence or for uses that are approved but without rigorous true placebo-controlled trials (namely vaccines) and/or with known safety concerns. That leaves us to speculate what the implications of this Assessment will be.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. Congress has given FDA the authority to require device manufacturers to provide cybersecurity information in their premarket submissions for a “cyber device.” Loose Ends IDEs.
This document is revised from a version published in October 2023. Firstly, leachables can compromise a products quality and therapeutic effect, by potentially interacting with the formulated drug product, the document reported. Moreover, manufacturers should documentinformation about their safety thresholds, the guidance stated.
The guidance published on the Medicines and Healthcare products Regulatory Agency (MHRA) website is broadly similar to arrangements laid out in a Brexit “no deal” document published last year. The concern is that UK patients will face delays to receiving the latest medicines because of the changes to the regulatory system.
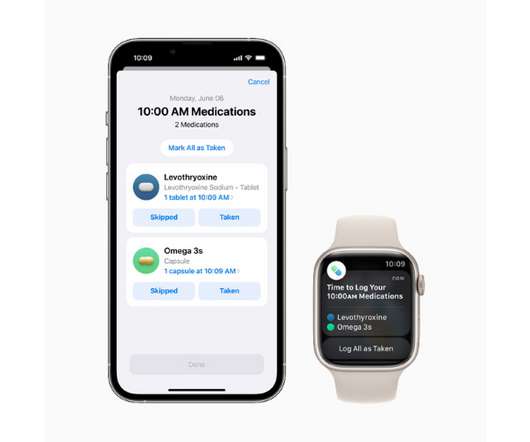
The new tool works as a component of Apple’s Health app and will let users add drugs or other health products like vitamins and supplements to a personal list – either by scanning a label or finding the product in a directory – and create custom schedules for them. New atrial fibrillation feature.
The document contains requirements and guidance for testing for bacterial endotoxins. This includes products that must be non-pyrogenic based on either intended use or non-pyrogenic label claim, or both. This document relating to microbiological methods is based on ANSI/AAMI ST72. Why is bacterial endotoxin testing important?
In other word, you need GMP documentation specially developed and used for your laboratory to guide you through everything you do in the lab. Depending on the objective of the lab, the need for GMP documentation will change for the laboratory. The significance of well-designed GMP documentations is immense.
Apply Data: Regulatory employees should be using stored data to intelligently create submission documents. By limiting documents full of free text fields and subjectivity companies can adopt a more digitised approach, where document templates can be compiled automatically from available data. Label Authoring and Tracking.
In fact, off-label drug uses can become widely entrenched in clinical practice and become predominant treatments for a given clinical condition. In a study published in JAMA Internal Medicine , off-label use lacking strong scientific evidence had a higher adverse drug event rate compared with on-label use.
Health Canada is seeking input from industry stakeholders on a new draft guidance document that discusses the use of electronic media in prescription drug labelling. Consultation on the draft guidance document is open until May 7, 2021. Guidelines for electronic labelling. Background and scope.
Issues with regulatory requirements and documentation can also cause significant delays, while any inaccuracies in translations on labelling can mean that dosage and storage information is not correctly understood or followed. And accurate labelling and translation are critical for this sector.
This can include everything from product testing and labelling to marketing and advertising requirements. As a result, nutraceutical startups need to stay informed about the latest regulatory changes and requirements in the states where they do business. Failure to do so can lead to legal and financial consequences.
Here, we take a look at the top five takeaways from the document: 1. Companies should still clearly document their reasons for supporting events, including virtual and hybrid international congresses, said the document. Identifying the appropriate code and label. The codes still apply. End of the host country.
The sampling strategy must be supported by sound and properly cited sources whose conclusions must be presented as supportive elements in the study documents. The following four elements of sampling methodology were found to be under-documented in RMM effectiveness studies: Supporting documentation for country/region selection.
By accurately documenting the movement of drugs, manufacturers can identify any quality issues that may arise during production, storage, or transportation. These systems enable real-time monitoring, accurate data capture, and seamless integration of information across various stakeholders, from manufacturers to healthcare providers.
ounces, but you must inform the officer at the checkpoint and they may need to inspect the container. It is best practice to carry these in a bottle with a prescription label. Doctor’s notes, prescriptions, or other documentation can be useful should your bags be inspected or if you need more medication while in transit.
FDA now seeks comments on this very program to support its continued collection of information as required under the Paperwork Reduction Act. On June 12, 2023, FDA issued a public notice to solicit comments on the information collection related to the voluntary submission of allegations of regulatory misconduct to CDRH.
Such non-device CDS are: (1) not intended to acquire, process, or analyse a medical image or a signal from an in vitro diagnostic device or a pattern or signal from a signal acquisition system (Criterion 1); (2) intended for the purpose of displaying, analysing, or printing medical information about a patient or other medical information (e.g.
The document contains detailed information on the suppliers and their product offerings, alongside contact details to aid your purchasing or hiring decision. Quality in terms of number of defective items, packaging and labelling, quality management system certification, research, development, and innovation.
The new draft guidance continues to borrow heavily from the NDA process and FDA notes that it used the same source material on which other drug application recommendations are based including the Common Technical Document (CTD). x 11-inch paper, and with hyperlinks to references and numbered pages.
To maintain the integrity and safety of these investigational products, you must adhere to Good Manufacturing Practice (GMP), FDA Clinical Trail Guidance Documents , and relevant ISO or EN standards. Additional documents included each month. Additional documents included each month. ensure traceability and compliance.
We organize all of the trending information in your field so you don't have to. Join 11,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content